Create a heatmap of correlation between receptors and transcription factors
Source:R/plot_fxns.R
cor_heatmap.RdCreates a heatmap of correlation values between receptors and transcription factors either with boolean threshold or with continuous values displayed
Usage
cor_heatmap(
dom,
bool = FALSE,
bool_thresh = 0.15,
title = TRUE,
feats = NULL,
recs = NULL,
mark_connections = FALSE,
...
)Arguments
- dom
Domino object with network built (
build_domino())- bool
Boolean indicating whether the heatmap should be continuous or boolean. If boolean then bool_thresh will be used to determine how to define activity as positive or negative.
- bool_thresh
Numeric indicating the threshold separating 'on' or 'off' for feature activity if making a boolean heatmap.
- title
Either a string to use as the title or a boolean describing whether to include a title. In order to pass the 'main' parameter to
ComplexHeatmap::Heatmap()you must set title to FALSE.- feats
Either a vector of features to include in the heatmap or 'all' for all features. If left NULL then the features selected for the signaling network will be shown.
- recs
Either a vector of receptors to include in the heatmap or 'all' for all receptors. If left NULL then the receptors selected in the signaling network connected to the features plotted will be shown.
- mark_connections
Boolean indicating whether to add an 'x' in cells where there is a connected receptor or TF. Default FALSE.
- ...
Other parameters to pass to
ComplexHeatmap::Heatmap(). Note that to use the 'main' parameter ofComplexHeatmap::Heatmap()you must set title = FALSE and to use 'annCol' or 'annColors' ann_cols must be FALSE.
See also
Heatmap Plotting Functions:
feat_heatmap(),
incoming_signaling_heatmap(),
signaling_heatmap()
Examples
example(build_domino, echo = FALSE)
#basic usage
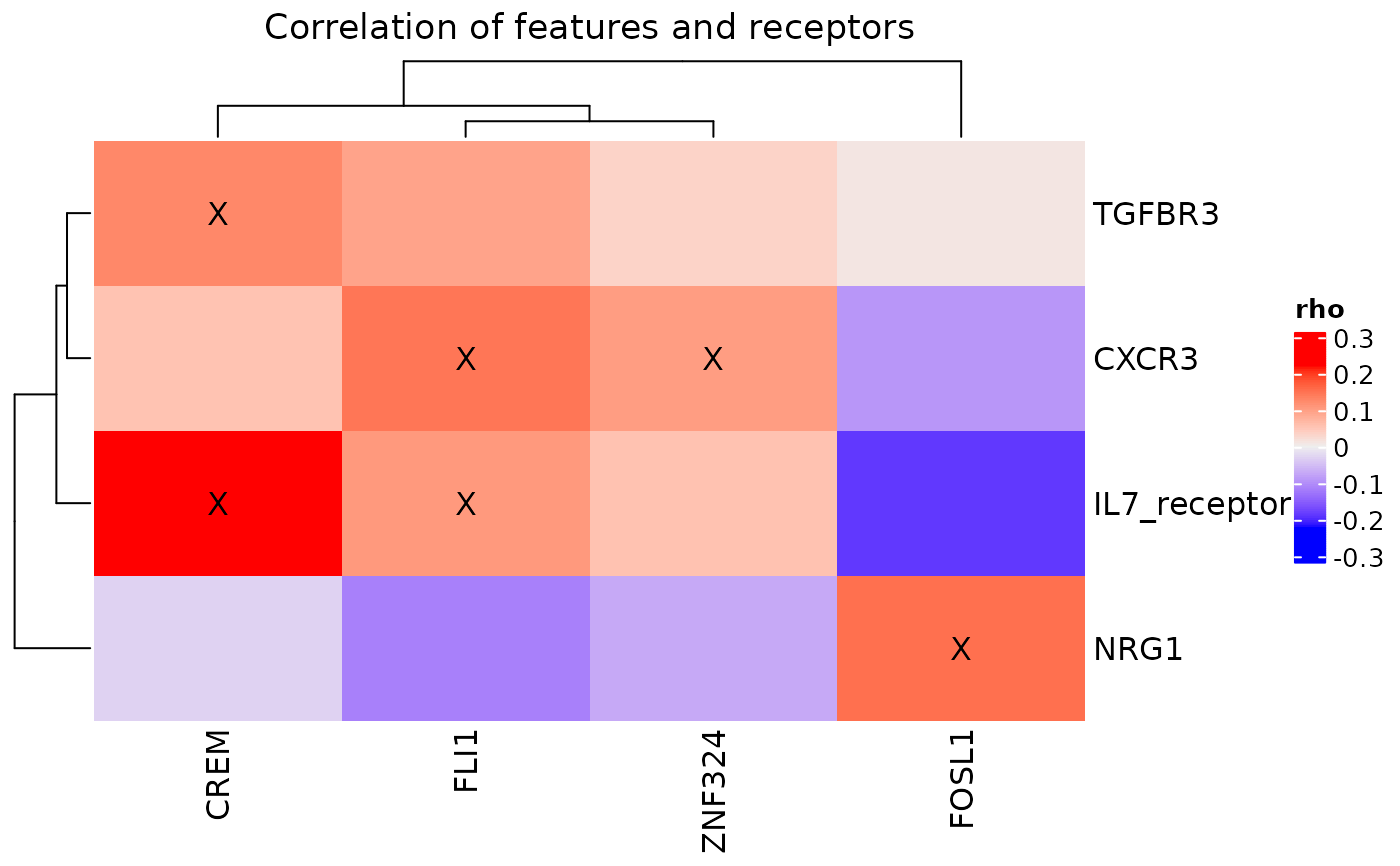
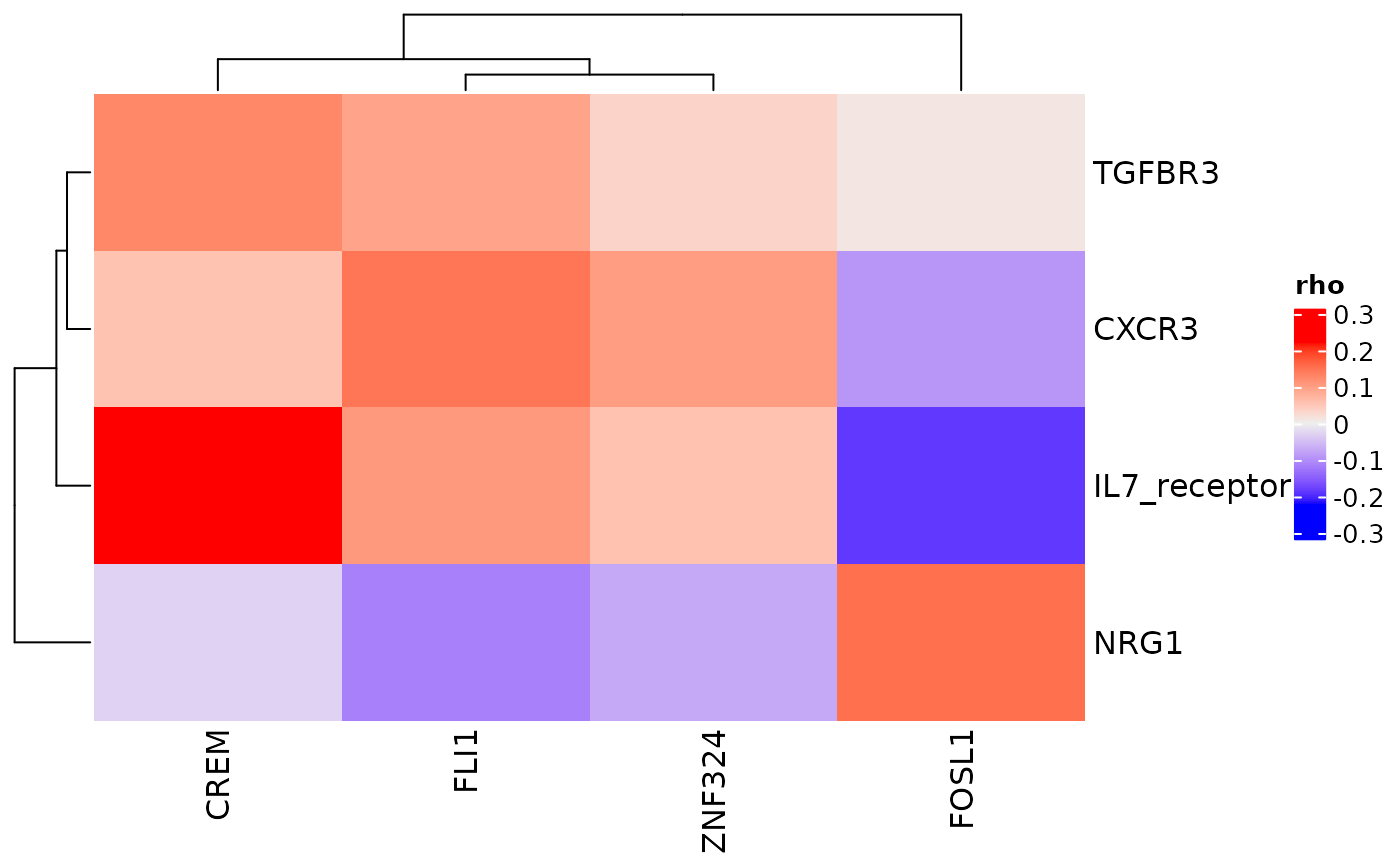
cor_heatmap(pbmc_dom_built_tiny, title = "PBMC R-TF Correlations")
 #show correlations above a specific value
cor_heatmap(pbmc_dom_built_tiny, bool = TRUE, bool_thresh = 0.1)
#show correlations above a specific value
cor_heatmap(pbmc_dom_built_tiny, bool = TRUE, bool_thresh = 0.1)
 #identify combinations that are connected
cor_heatmap(pbmc_dom_built_tiny, bool = FALSE, mark_connections = TRUE)
#identify combinations that are connected
cor_heatmap(pbmc_dom_built_tiny, bool = FALSE, mark_connections = TRUE)