dominoSignal is a tool for analysis of intra- and intercellular signaling in single cell RNA sequencing (scRNAseq) data based on transcription factor (TF) activation. Here, we show a basic example pipeline for generating communication networks as a domino object. This vignette will demonstrate how to use dominoSignal on the 10X Genomics Peripheral Blood Mononuclear Cells (PBMC) data set of 2,700 cells. The data can be downloaded here.
Options and Setup
Libraries and set up
Data used in our vignettes can be downloaded from Zenodo.
# BiocFileCache helps with managing files across sessions
bfc <- BiocFileCache::BiocFileCache(ask = FALSE)
data_url <- "https://zenodo.org/records/10951634/files"
# download the reduced pbmc files preprocessed PBMC 3K scRNAseq data set as a
# Seurat object
pbmc_url <- paste0(data_url, "/pbmc3k_sce.rds")
pbmc <- BiocFileCache::bfcrpath(bfc, pbmc_url)
# download scenic results
scenic_auc_url <- paste0(data_url, "/auc_pbmc_3k.csv")
scenic_auc <- BiocFileCache::bfcrpath(bfc, scenic_auc_url)
scenic_regulon_url <- paste0(data_url, "/regulons_pbmc_3k.csv")
scenic_regulon <- BiocFileCache::bfcrpath(bfc, scenic_regulon_url)
# download CellPhoneDB files
cellphone_url <- "https://github.com/ventolab/cellphonedb-data/archive/refs/tags/v4.0.0.tar.gz"
cellphone_tar <- BiocFileCache::bfcrpath(bfc, cellphone_url)
cellphone_dir <- paste0(tempdir(), "/cellphone")
untar(tarfile = cellphone_tar, exdir = cellphone_dir)
cellphone_data <- paste0(cellphone_dir, "/cellphonedb-data-4.0.0/data")
# directory for created inputs to pySCENIC and dominoSignal
input_dir <- paste0(tempdir(), "/inputs")
if (!dir.exists(input_dir)) {
dir.create(input_dir)
}Data preparation
Analysis of cell-cell communication with dominoSignal often follows initial processing and annotation of scRNAseq data. We used the OSCA workflow to prepare the data set for analysis with dominoSignal. The complete processing script is available in the data-raw directory of the dominoSignal package. The processed data can be downloaded from Zenodo.
pbmc <- readRDS(pbmc)Installation
Installation of dominoSignal from Bioconductor can be achieved using the BiocManager package.
if (!require("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("dominoSignal")Loading TF and R - L data
Loading SCENIC Results
The TF activities are a required input for dominoSignal, and the regulons learned by SCENIC are an important but optional input needed to annotate TF-target interactions and to prune TF-receptor linkages where the receptor is a target of the TF. This prevents the distinction of receptor expression driving TF activity or the TF inducing the receptor’s expression.
regulons <- read.csv(scenic_regulon)
auc <- read.table(scenic_auc, header = TRUE, row.names = 1, stringsAsFactors = FALSE,
sep = ",")The initial regulons data frame read into R from the ctx function has
two rows of column names that need to be replaced with one succinct
description. dominoSignal has changed the input format for TF regulons
to be a list storing vectors of target genes in each regulon, where the
names of the list are the TF genes. This facilitates the use of
alternative methods for TF activity quantification. We provide a helper
function, create_regulon_list_scenic() for easy retrieval
of TF regulons from the output of the pySCENIC ctx
function.
regulons <- regulons[-1:-2, ]
colnames(regulons) <- c("TF", "MotifID", "AUC", "NES", "MotifSimilarityQvalue", "OrthologousIdentity",
"Annotation", "Context", "TargetGenes", "RankAtMax")
regulon_list <- create_regulon_list_scenic(regulons = regulons)Users should be aware that the AUC matrix from SCENIC is loaded in a cell x TF
orientation and should be transposed to TF x cell orientation. pySCENIC
also appends “(+)” to TF names that are converted to “…” upon loading
into R. These characters can be included without affecting the results
of dominoSignal analysis but can be confusing when querying TF features
in the data. We recommend comprehensive removal of the “…” characters
using the gsub() function.
auc_in <- as.data.frame(t(auc))
# Remove pattern '...' from the end of all rownames:
rownames(auc_in) <- gsub("\\.\\.\\.$", "", rownames(auc_in))Load CellPhoneDB Database
dominoSignal has been updated to read ligand - receptor data bases in a uniform data frame format referred to as a receptor-ligand map (rl_map) to enable the use of alternative or updated reference databases in addition to the particular version of CellPhoneDB’s database in older versions of Domino. Each row corresponds to a ligand - receptor interaction. Genes participating in an interaction are referred to as “partner A” and “partner B” without requiring for fixed ordering of whether A or B is the ligand and vice versa. The minimum required columns of this data frame are:
- int_pair: the names of the interacting ligand and receptor separated by ” & ”
- gene_A: the gene or genes encoding partner A
- gene_B: the gene or genes encoding partner B
- type_A: (“L”, “R”) - indicates whether partner A is a ligand (“L”) or receptor (“R”)
- type_B: (“L”, “R”) - indicates whether partner B is a ligand (“L”) or receptor (“R”)
Additional annotation columns can be provided such as name_A and name_B for ligands or receptors whose name in the interaction database does not match the names of their encoding genes. This formatting also allows for the consideration of ligand and receptor complexes comprised of a heteromeric combination of multiple proteins that must be co-expressed to function. In these cases, the “name_*” column shows the name of the protein complex, and the “gene_*” column shows the names of the genes encoding the components of the complex separated by commas “,”. When plotting results from the build domino object, the names of the interacting ligands and receptors will be used based on combined expression of the complex components.
To facilitate the use of this formatting with the CellPhoneDB
database, we include a helper function,
create_rl_map_cellphonedb(), that automatically parses
files from the CellPhoneDB database to arrive at the rl_map format.
complexes <- read.csv(paste0(cellphone_data, "/complex_input.csv"), stringsAsFactors = FALSE)
genes <- read.csv(paste0(cellphone_data, "/gene_input.csv"), stringsAsFactors = FALSE)
interactions <- read.csv(paste0(cellphone_data, "/interaction_input.csv"), stringsAsFactors = FALSE)
proteins <- read.csv(paste0(cellphone_data, "/protein_input.csv"), stringsAsFactors = FALSE)
rl_map <- create_rl_map_cellphonedb(genes = genes, proteins = proteins, interactions = interactions,
complexes = complexes, database_name = "CellPhoneDB_v4.0" # database version used
)
knitr::kable(head(rl_map))| int_pair | name_A | uniprot_A | gene_A | type_A | name_B | uniprot_B | gene_B | type_B | annotation_strategy | source | database_name | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | RAreceptor_RXRG & atRetinoicAcid_byALDH1A3 | RAreceptor_RXRG | P48443,P29373 | RXRG,CRABP2 | R | atRetinoicAcid_byALDH1A3 | P47895 | ALDH1A3 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 5 | RAreceptor_RXRG & atRetinoicAcid_byALDH1A2 | RAreceptor_RXRG | P48443,P29373 | RXRG,CRABP2 | R | atRetinoicAcid_byALDH1A2 | O94788 | ALDH1A2 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 6 | RAreceptor_RXRG & atRetinoicAcid_byALDH1A1 | RAreceptor_RXRG | P48443,P29373 | RXRG,CRABP2 | R | atRetinoicAcid_byALDH1A1 | P00352 | ALDH1A1 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 7 | RAreceptor_RXRB & atRetinoicAcid_byALDH1A3 | RAreceptor_RXRB | P28702,P29373 | RXRB,CRABP2 | R | atRetinoicAcid_byALDH1A3 | P47895 | ALDH1A3 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 8 | RAreceptor_RXRB & atRetinoicAcid_byALDH1A2 | RAreceptor_RXRB | P28702,P29373 | RXRB,CRABP2 | R | atRetinoicAcid_byALDH1A2 | O94788 | ALDH1A2 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 9 | RAreceptor_RXRB & atRetinoicAcid_byALDH1A1 | RAreceptor_RXRB | P28702,P29373 | RXRB,CRABP2 | R | atRetinoicAcid_byALDH1A1 | P00352 | ALDH1A1 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
Optional: Adding interactions manually
The change to use rl_map formatting also enables users to manually
append interactions of interest that are not included in the interaction
database if need be. This can be attained by formatting the desired
interactions as a data frame with the same column headers as the rl_map
and using the rbind() function.
# Integrin complexes are not annotated as receptors in CellPhoneDB_v4.0
# collagen-integrin interactions between cells may be missed unless tables from
# the CellPhoneDB reference are edited or the interactions are manually added
col_int_df <- data.frame(int_pair = "a11b1 complex & COLA1_HUMAN", name_A = "a11b1 complex",
uniprot_A = "P05556,Q9UKX5", gene_A = "ITB1,ITA11", type_A = "R", name_B = "COLA1_HUMAN",
uniprot_B = "P02452,P08123", gene_B = "COL1A1,COL1A2", type_B = "L", annotation_strategy = "manual",
source = "manual", database_name = "manual")
rl_map_append <- rbind(col_int_df, rl_map)
knitr::kable(head(rl_map_append))| int_pair | name_A | uniprot_A | gene_A | type_A | name_B | uniprot_B | gene_B | type_B | annotation_strategy | source | database_name | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | a11b1 complex & COLA1_HUMAN | a11b1 complex | P05556,Q9UKX5 | ITB1,ITA11 | R | COLA1_HUMAN | P02452,P08123 | COL1A1,COL1A2 | L | manual | manual | manual |
| 4 | RAreceptor_RXRG & atRetinoicAcid_byALDH1A3 | RAreceptor_RXRG | P48443,P29373 | RXRG,CRABP2 | R | atRetinoicAcid_byALDH1A3 | P47895 | ALDH1A3 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 5 | RAreceptor_RXRG & atRetinoicAcid_byALDH1A2 | RAreceptor_RXRG | P48443,P29373 | RXRG,CRABP2 | R | atRetinoicAcid_byALDH1A2 | O94788 | ALDH1A2 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 6 | RAreceptor_RXRG & atRetinoicAcid_byALDH1A1 | RAreceptor_RXRG | P48443,P29373 | RXRG,CRABP2 | R | atRetinoicAcid_byALDH1A1 | P00352 | ALDH1A1 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 7 | RAreceptor_RXRB & atRetinoicAcid_byALDH1A3 | RAreceptor_RXRB | P28702,P29373 | RXRB,CRABP2 | R | atRetinoicAcid_byALDH1A3 | P47895 | ALDH1A3 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
| 8 | RAreceptor_RXRB & atRetinoicAcid_byALDH1A2 | RAreceptor_RXRB | P28702,P29373 | RXRB,CRABP2 | R | atRetinoicAcid_byALDH1A2 | O94788 | ALDH1A2 | L | curated | uniprot;reactome | CellPhoneDB_v4.0 |
Analysis with Domino object
dominoSignal analysis takes place in two steps.
-
initializes the domino result object
assesses differential TF activity across cell clusters by Wilcoxon rank-sum test
establishes TF-receptor linkages based on Spearman correlation of TF activities with receptor expression across the queried data set.
-
sets the parameters for which TFs and receptors are called as active within a cell cluster
aggregates the scaled expression of ligands capable of interacting with the active receptors for assessment of ligand type and cellular source triggering activation of a receptor.
Required inputs from data set
dominoSignal infers active receipt of signals via receptors based on
the correlation of the receptor’s expression with TF activity across the
data set and differential activity of the TF within a cell cluster.
Correlations are conducted using scaled expression values rather than
raw counts or normalized counts. For assessment of receptor activity on
a per cell type basis, a named vector of cell cluster assignments, where
the names are cell barcodes matching the expression matrix, are
provided. Assessing signaling based on other categorical groupings of
cells can be achieved by passing these groupings as “clusters” to
build_domino() in place of cell types. dominoSignal accepts
a matrix of counts, a matrix of scaled counts, and a
named vector of cell cluster labels as factors. Shown
below is how to extract these elements from a SingleCellExperiment
object. Note that since the data is scaled by gene, genes with no
expression in any cell need to be removed.
counts = assay(pbmc, "counts")
logcounts = assay(pbmc, "logcounts")
logcounts = logcounts[rowSums(logcounts) > 0, ]
z_scores = t(scale(t(logcounts)))
clusters = factor(pbmc$cell_type)
names(clusters) = colnames(pbmc)Note: Ligand and receptor expression can only be assessed for genes included in the z_scores matrix. Many scRNAseq analysis pipelines recommend storing only genes with high variance in scaled expression slots for these data objects, thereby missing many genes encoding ligands and receptors. Ensure that all your genes of interest are included in the rows of your z_scores matrix. Scaled expression was calculated for all genes in this PBMC data set after removal of genes expressed in less than three cells.
Create Domino object
At this point, the create_domino() function can be used
to make the object. Parameters of note include “use_clusters” which is
required to assess signaling between cell types rather than linkage of
TFs and receptors broadly across the data set. “use_complexes” decides
if receptors that function in heteromeric complexes will be considered
in testing linkages between TFs and receptors. If TRUE, a receptor
complex is only linked to a TF if a majority of the component genes meet
the Spearman correlation threshold. “remove_rec_dropout” decides whether
receptor values of zero will be considered in correlation calculations;
this measure is intended to reduce the effect of dropout on receptors
with low expression.
To run create_domino() with matrix and vector
inputs:
pbmc_dom <- create_domino(rl_map = rl_map, features = auc_in, counts = counts, z_scores = z_scores,
clusters = clusters, tf_targets = regulon_list, use_clusters = TRUE, use_complexes = TRUE,
remove_rec_dropout = FALSE)
# rl_map: receptor - ligand map data frame features: TF activation scores (AUC
# matrix) counts: counts matrix z_scores: scaled expression data clusters:
# named vector of cell cluster assignments tf_targets: list of TFs and their
# regulons use_clusters: assess receptor activation and ligand expression on a
# per-cluster basis use_complexes: include receptors and genes that function as
# a complex in results remove_rec_dropout: whether to remove zeroes from
# correlation calculationsFor information on what is stored in a domino object and how to access it, please see our vignette on the structure of domino objects.
Build Domino Network
build_domino() finalizes the construction of the domino
object by setting parameters for identifying TFs with differential
activation between clusters, receptor linkage with TFs based on
magnitude of positive correlation, and the minimum percentage of cells
within a cluster that have expression of a receptor for the receptor to
be called as active.
There are also options for thresholds of the number of TFs that may be called active in a cluster and the number of receptors that may be linked to any one TF. For thresholds of n TFs and m receptors, the bottom n TFs by lowest p-values from the Wilcoxon rank sum test and the top m receptors by Spearman correlation coefficient are chosen.
pbmc_dom <- build_domino(dom = pbmc_dom, min_tf_pval = 0.001, max_tf_per_clust = 25,
max_rec_per_tf = 25, rec_tf_cor_threshold = 0.25, min_rec_percentage = 0.1)
# min_tf_pval: Threshold for p-value of DE for TFs rec_tf_cor_threshold:
# Minimum correlation between receptor and TF min_rec_percentage: Minimum
# percent of cells that must express receptorThresholds for both the number of receptors and TFs can be sent to infinity (Inf) to collect all receptors and TFs that meet other thresholds.
pbmc_dom_all <- build_domino(dom = pbmc_dom, min_tf_pval = 0.001, max_tf_per_clust = Inf,
max_rec_per_tf = Inf, rec_tf_cor_threshold = 0.25, min_rec_percentage = 0.1)Visualization of Domino Results
Multiple functions are available to visualize the intracellular networks between receptors and TFs and the ligand - receptor mediated intercellular networks between cell types.
Summarize TF Activity and Linkage
Enrichment of TF activities by cell types can be visualized by
feat_heatmap() which plots data set-wide TF activity scores
as a heatmap.
feat_heatmap(pbmc_dom, norm = TRUE, bool = FALSE, use_raster = FALSE, row_names_gp = grid::gpar(fontsize = 4))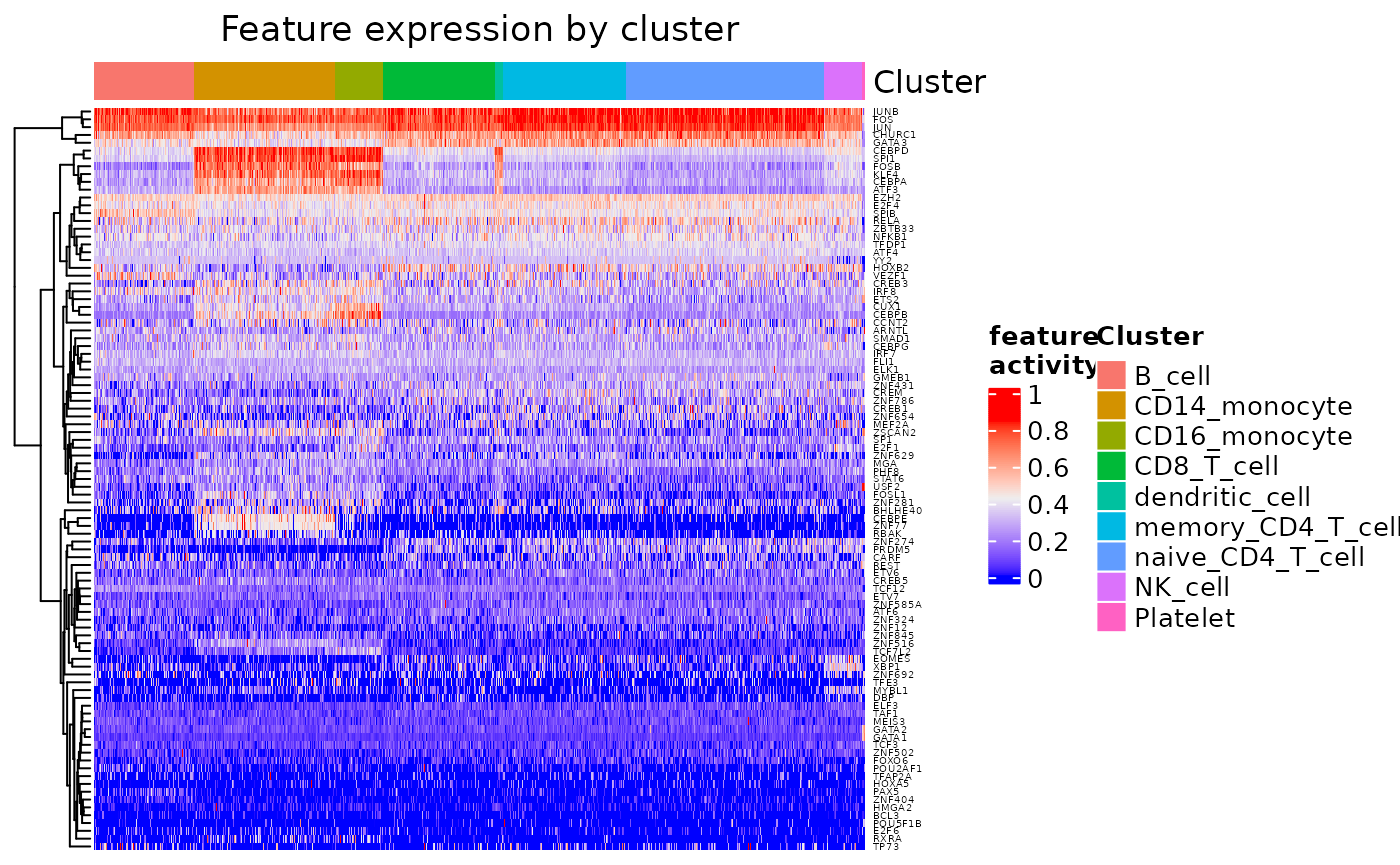
Cumulative signaling between cell types
The cumulative degree of signaling between clusters is assessed as
the sum of the scaled expression of ligands targeting active receptors
on another cluster. This can be visualized in graph format using the
signaling_network() function. Nodes represent each cell
cluster and edges scale with the magnitude of signaling between the
clusters. The color of the edge corresponds to the sender cluster for
that signal.
signaling_network(pbmc_dom, edge_weight = 0.5, max_thresh = 3)
Signaling networks can also be drawn with the edges only rendering the signals directed towards a given cell type or signals from one cell type directed to others. To see those options in use, as well as other plotting functions and their options, please see our plotting vignette.
Specific Signaling Interactions between Clusters
Beyond the aggregated degree of signaling between cell types, the
degrees of signaling through specific ligand - receptor interactions can
be assessed. gene_network() provides a graph of linkages
between active TFs in a cluster, the linked receptors in that cluster,
and the possible ligands of these active receptors.
gene_network(pbmc_dom, clust = "dendritic_cell", layout = "grid")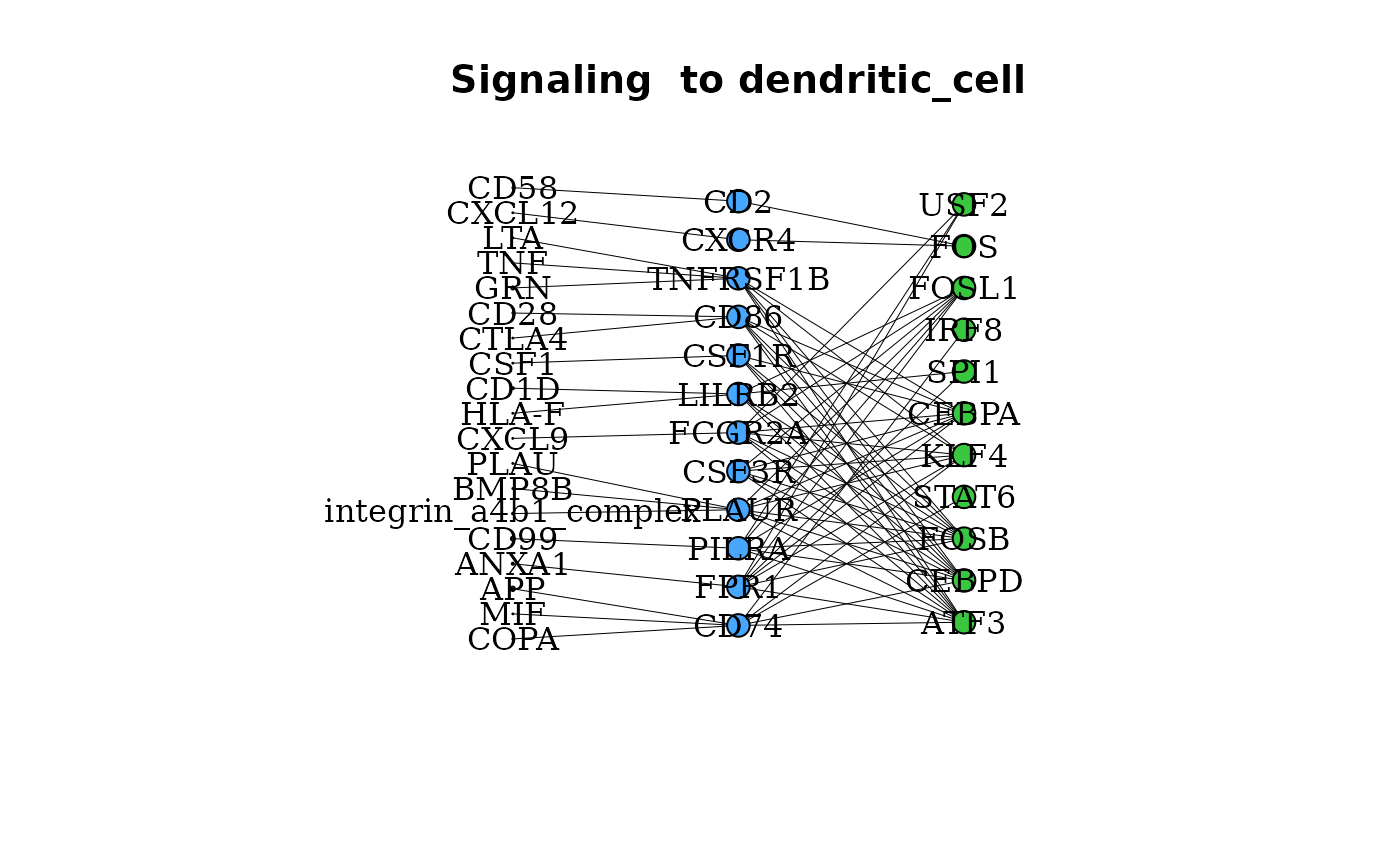
New to dominoSignal, gene_network() can be used between
two clusters to determine if any of the possible ligands for a given
receptor are expressed by a putative outgoing signaling cluster.
gene_network(pbmc_dom, clust = "dendritic_cell", OutgoingSignalingClust = "CD14_monocyte",
layout = "grid")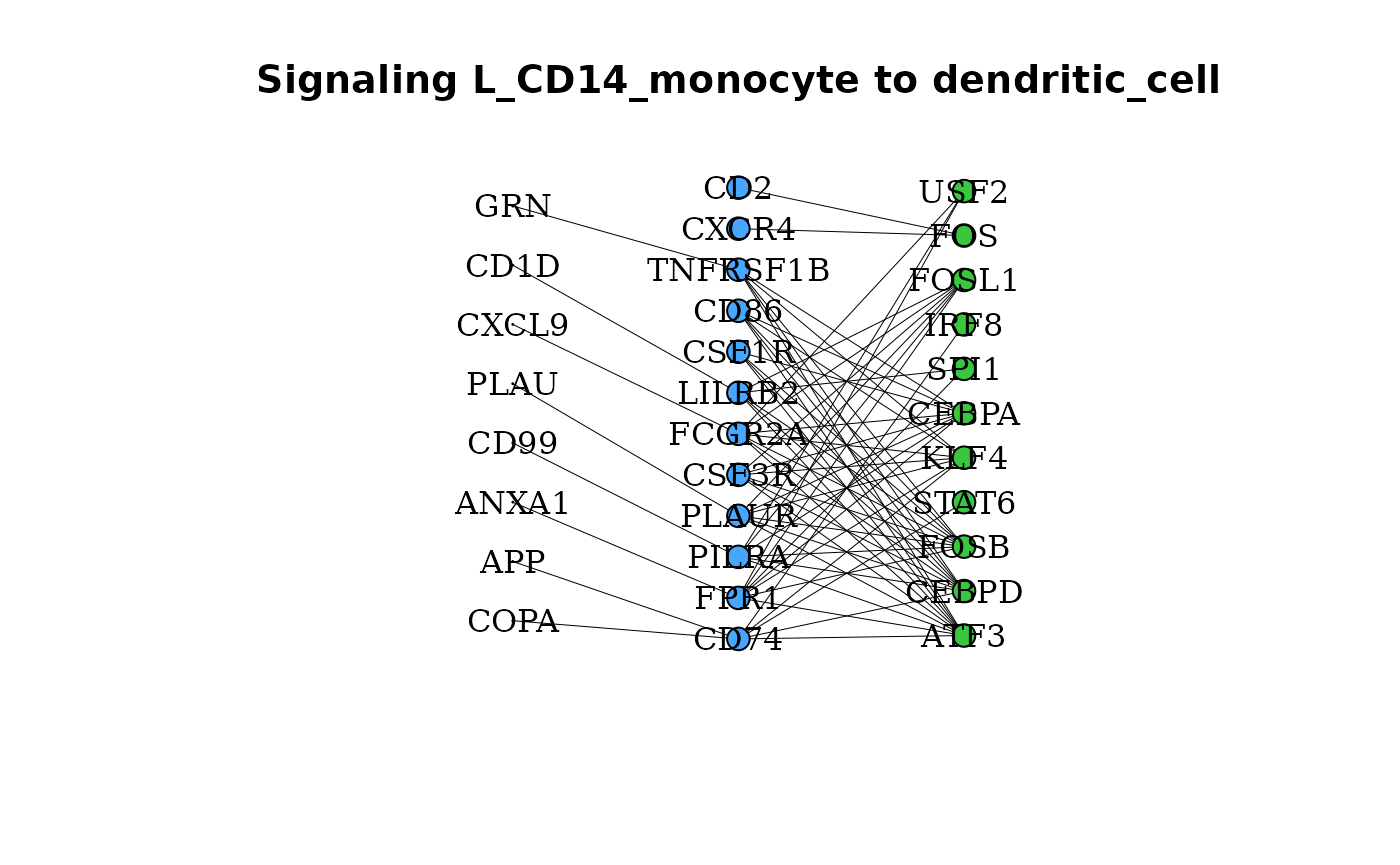
A comprehensive assessment of ligand expression targeting active
receptors on a given cluster can be assessed with
incoming_signaling_heatmap().
incoming_signaling_heatmap(pbmc_dom, rec_clust = "dendritic_cell", max_thresh = 2.5,
use_raster = FALSE)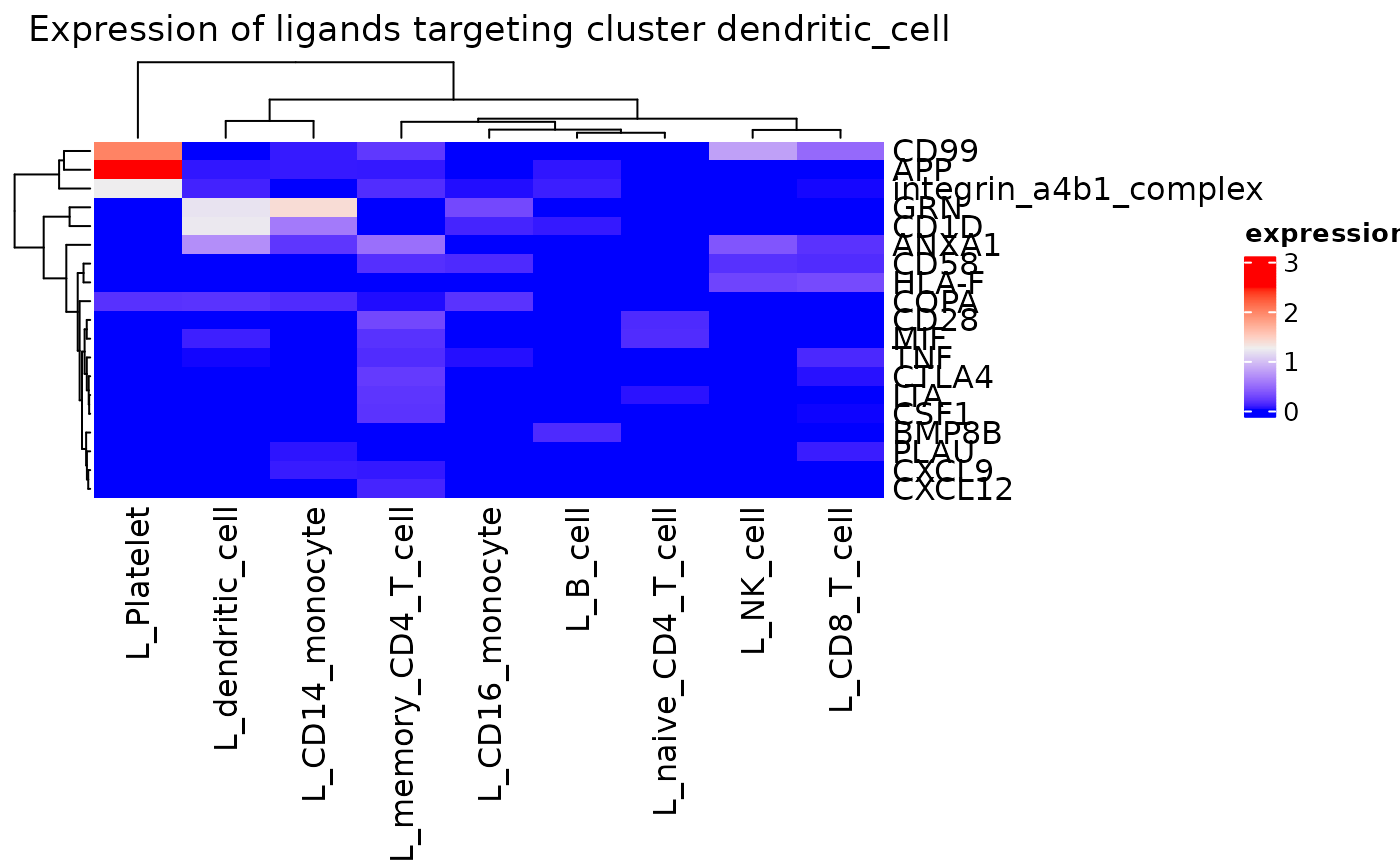
Another form of comprehensive ligand expression assessment is available for individual active receptors in the form of circos plots new to dominoSignal. The outer arcs correspond to clusters in the domino object with inner arcs representing each possible ligand of the plotted receptor. Arcs are drawn between ligands on a cell type and the receptor if the ligand is expressed above the specified threshold. Arc widths correspond to the mean express of the ligand by the cluster with the widest arc width scaling to the maximum expression of the ligand within the data.
circos_ligand_receptor(pbmc_dom, receptor = "CD74")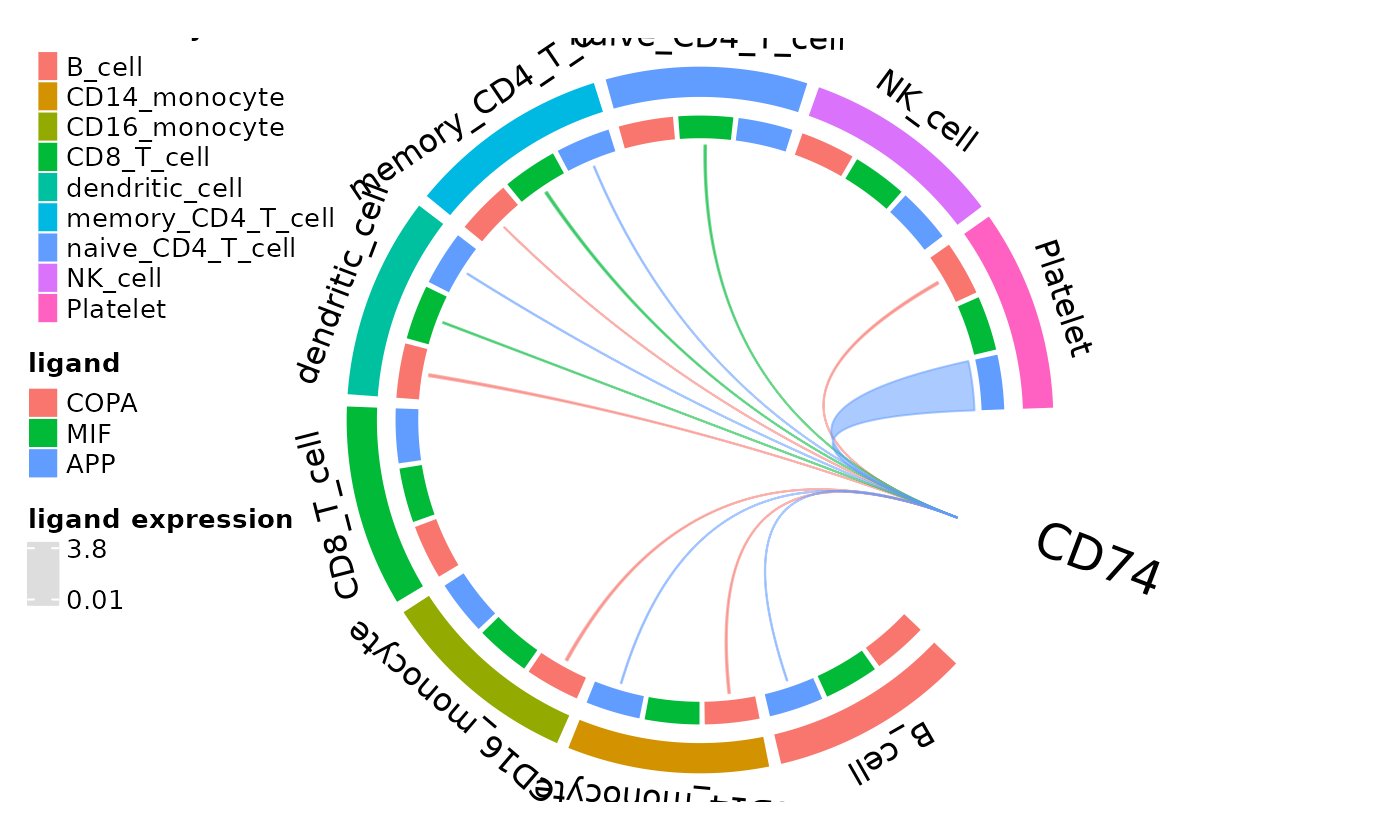
Continued Development
Since dominoSignal is a package still being developed, there are new functions and features that will be implemented in future versions. In the meantime, we have put together further information on plotting and the domino object structure if you would like to explore more of the package’s functionality. Additionally, if you find any bugs, have further questions, or want to share an idea, please let us know here.
Vignette Build Information
Date last built and session information:
Sys.Date()
#> [1] "2026-05-05"
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] grid stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] knitr_1.51 ComplexHeatmap_2.28.0
#> [3] circlize_0.4.18 plyr_1.8.9
#> [5] SingleCellExperiment_1.34.0 SummarizedExperiment_1.42.0
#> [7] Biobase_2.72.0 GenomicRanges_1.64.0
#> [9] Seqinfo_1.2.0 IRanges_2.46.0
#> [11] S4Vectors_0.50.0 BiocGenerics_0.58.0
#> [13] generics_0.1.4 MatrixGenerics_1.24.0
#> [15] matrixStats_1.5.0 dominoSignal_1.6.0
#>
#> loaded via a namespace (and not attached):
#> [1] DBI_1.3.0 httr2_1.2.2 formatR_1.14
#> [4] biomaRt_2.68.0 rlang_1.2.0 magrittr_2.0.5
#> [7] clue_0.3-68 GetoptLong_1.1.1 otel_0.2.0
#> [10] compiler_4.6.0 RSQLite_2.4.6 png_0.1-9
#> [13] systemfonts_1.3.2 vctrs_0.7.3 stringr_1.6.0
#> [16] pkgconfig_2.0.3 shape_1.4.6.1 crayon_1.5.3
#> [19] fastmap_1.2.0 backports_1.5.1 dbplyr_2.5.2
#> [22] XVector_0.52.0 rmarkdown_2.31 ragg_1.5.2
#> [25] purrr_1.2.2 bit_4.6.0 xfun_0.57
#> [28] cachem_1.1.0 jsonlite_2.0.0 progress_1.2.3
#> [31] blob_1.3.0 DelayedArray_0.38.1 broom_1.0.12
#> [34] parallel_4.6.0 prettyunits_1.2.0 cluster_2.1.8.2
#> [37] R6_2.6.1 bslib_0.10.0 stringi_1.8.7
#> [40] RColorBrewer_1.1-3 car_3.1-5 jquerylib_0.1.4
#> [43] Rcpp_1.1.1-1.1 iterators_1.0.14 Matrix_1.7-5
#> [46] igraph_2.3.1 tidyselect_1.2.1 abind_1.4-8
#> [49] yaml_2.3.12 doParallel_1.0.17 codetools_0.2-20
#> [52] curl_7.1.0 lattice_0.22-9 tibble_3.3.1
#> [55] withr_3.0.2 KEGGREST_1.52.0 S7_0.2.2
#> [58] evaluate_1.0.5 desc_1.4.3 BiocFileCache_3.2.0
#> [61] Biostrings_2.80.0 pillar_1.11.1 ggpubr_0.6.3
#> [64] filelock_1.0.3 carData_3.0-6 foreach_1.5.2
#> [67] hms_1.1.4 ggplot2_4.0.3 scales_1.4.0
#> [70] glue_1.8.1 tools_4.6.0 ggsignif_0.6.4
#> [73] fs_2.1.0 tidyr_1.3.2 AnnotationDbi_1.74.0
#> [76] colorspace_2.1-2 Formula_1.2-5 cli_3.6.6
#> [79] rappdirs_0.3.4 textshaping_1.0.5 S4Arrays_1.12.0
#> [82] dplyr_1.2.1 gtable_0.3.6 rstatix_0.7.3
#> [85] sass_0.4.10 digest_0.6.39 SparseArray_1.12.2
#> [88] rjson_0.2.23 htmlwidgets_1.6.4 farver_2.1.2
#> [91] memoise_2.0.1 htmltools_0.5.9 pkgdown_2.2.0
#> [94] lifecycle_1.0.5 httr_1.4.8 GlobalOptions_0.1.4
#> [97] bit64_4.8.0